This blog post is spurred on by some
interesting conversations happening online about effective teaching and lecture
prep strategies, so I though I would write something about my experiences over
the last 8 years with teaching a large intro biology course, with the hope that
some of this will be of use to others.
Our story begins in the Winter of 2011, my
first semester teaching BI111 (Biological Diversity and Evolution), the 2nd
half of the first-year biology courses (BI110, which runs in the Fall & focuses
on Cells+Molecular Biology). BI111 has a broad course description (“Interactions
of organisms with each other and with the environment in the ongoing process of
evolution by natural selection are examined in the context of the interplay of
form with function…”), which can be interpreted in any number of ways. I should
also point out that I teach two sections of this course (in those days it was
~250-300 students in each, but now each section is ~450 students), and that
while most students are in Science, the majority are NOT Biology majors (with the most common non-biology majors Kin,
Health Science, Math, Chem, Psychology etc…), and as a result of all these
different paths, not everyone in my class has taken grade 12 biology. So there
is a wide range of prior experiences and reasons for taking this class.
As I mentioned, this class has a broad
mandate, and the mistake I made in my first year was thinking that I should try
and cover as much in the 2nd half of the intro bio textbook that I
could. This sentiment is common in 1st year classes, with the idea
being that you need to prepare students for any and all types of biology they
may encounter in their second year. This is an obvious mistake, and quickly
became apparent to me, as I tried to cram 2-3 chapters’ worth of topics into
each week’s lectures (3 x 50min lectures/week x 12 weeks). Not only was I
floundering at keeping up with the lecture prep, and because of the sheer
amount of content, I couldn’t delve deep into any one topic during lectures.
The course had already adopted the use of iClicker personal response devices,
but my major use of them was for strictly ‘definitional’ questions (Which of
the following best describes “term X”), and did not really test anyone’s
knowledge. The whole semester was
nothing but stress and frustration, and the less said about it the better.
So at the end of this first semester I
decided that I needed to fundamentally change up teaching strategy for this
course. It is worth mentioning at this point, that the large number of changes
I ended up adopting were not done all during the next year, but built up over
time. This has only been achievable, as I have had the luxury of having a
consistent yearly teaching assignment while at Laurier, allowing me to the time
and opportunity to better develop each of the new elements. The first thing out
the window was the “try and teach the totality of human knowledge” idea.
Instead I decided to structure the class in such a way that I focused on fewer
topics in a given week, and tried to make the topics covered in different weeks
connect with each other. At the same time I made a conscious choice to move
away from spending lectures defining terms, and instead use that time to apply
the concepts associated with the definitions. This requires students to come
into the classroom each week with at least a working knowledge of the key
terms/ideas of the lecture, which meant that students should have read over the
relevant textbook passages beforehand. This is an ongoing challenge for most profs,
but I think I have solved it through the combined use of “Learning Objective”
(LO) documents and “Entrance/Exit Quizzes”, which I will discuss below.
One of the biggest (logical & rational)
concerns of an undergrad student is that they understand what they are expected
to know in a given course. In an intro course, which covers many
topics/chapters, this can be a big concern. So when I was redesigning the
course, the first thing I did was go through my chosen textbook chapters (and
other readings – more on that below), and create a Learning Objective Document,
which is shared with the students at the start of the semester. This document
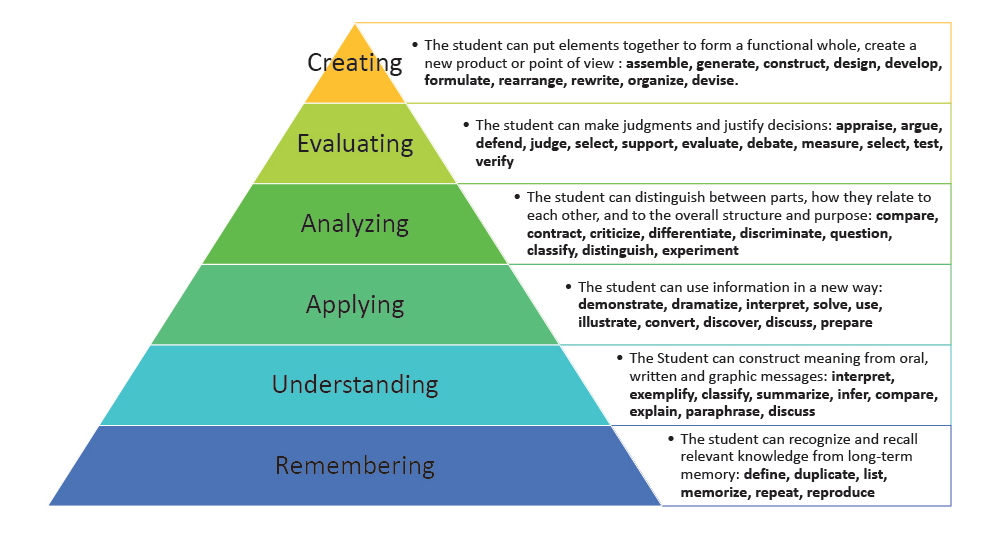
has two sections (BASIC and ADVANCED), and are framed using Bloom’s Taxonomy
words (you can see an example here: https://cpb-ap-se2.wpmucdn.com/global2.vic.edu.au/dist/d/8496/files/2015/09/Screen-Shot-2015-09-05-at-9.22.41-am-1jq2xw2.png,
and another here: https://wp0.vanderbilt.edu/cft/guides-sub-pages/blooms-taxonomy
). The basic section focuses primarily on defining
the terms I would expect everyone to be familiar with. The textbook does a
great job at providing simple descriptions of concepts, and it would be
redundant to spend lecture time repeating it. To ensure the students are
familiar with these terms, on the weekend before the start of every week’s
lectures I have a very simple online CMS-administered multiple-choice “Entrance
Quiz” of ~8-10 questions based explicitly on the terms listed in the basic
section of the Learning Objective document. This is a “low-stakes” quiz (not
worth a large % of the course grade, but not very challenging) that ends up having high participation (~90%
on any given week), that also allows me to see (before the week’s lectures
begin) if there are any terms that are tripping up students. Since lecture time
is (largely) freed-up from going over simple definitions, this gives me more
time to explore the week’s topic in more depth. This involves going into more
detail than in the textbook, and apply their understanding of the definitions
to new situations/contexts. These are more challenging tasks, and are mirrored
in the “Advanced” section of that week’s lecture and focus on
describing/explaining/discussing/predicting. At the end of each week I open up
an online 8-10 multiple choice “Exit Quiz” that focuses on these advanced
topics that runs over the weekend (and concurrently with the opening of the
next week’s “Entrance Quiz). I have also recently started incorporating
primarily literature reading into my course readings as the ability to read
scientific papers is a skill that students will need in their future studies.
For ~each week I have chosen a paper published in the journal Biology Letters,
which have very short-form manuscripts (often 2-3 pages max) that are directly
tied into the topic being covered in that week’s lectures (eg. 1, 2, 3), and can easily be swapped between years. An entrance quiz
question may just focus on some simple term defined in the introduction or
methods. During the week we would discuss the study in class, and an exit quiz
question may be based on that discussion, or an extension of the experiment/study.
Furthermore, I use (*and * tell my students that I use) the LO sheets when composing
my mid-term and final exam questions, so that there are both short-term and
long-term benefits of using them.
Now I want to emphasize that each of these
elements takes time to construct, and can be a bit overwhelming if you were
going to try and do it all at once. I think it would be perfectly fine if you
developed the LOs in the 1st year, and then bring entrance and exit
quizzes online in subsequent year(s).
So now that I’d freed up large chunks of
lecture time, what to with it? Following the principle of ‘less is more’ I try
to focus my time exploring on one or two key ideas in depth. Textbooks examples
and definitions are pretty clear-cut, the result of lots of distilling and
generalizing. I often try to show my students through case studies that things
are not as simple. If we are talking about species concepts, I’ll go over the
pros and cons of various concepts, and then present them with examples where
(depending on the method used) they’ll end up with different conclusions. When
discussing the general effects associated with plant hormones, I’ll ask them to
hypothesize the likely changes in expression that have arisen between the
ancestral and derived Brassica oleracea cultivars (broccoli, kale, kohlrabi
etc..). I also try to use the lecture time to present physical examples of the
lecture’s subjects. My classroom is equipped with a document camera, which is
great because I can, for example, demonstrate to the students through
dissections of flowers the various vegetative and non-vegetative whorls and
structures, or by weighing slices of sweet potato that have been soaking in
hypotonic or hypertonic solutions, how water moves across the plant cell
membranes. I also use active learning exercises to illustrate how
organisms/populations/communities change over time, with students playing
different roles. Using iClickers and (lots of) playing cards, I can demonstrate the Hardy-Weinberg principle, and how violations of its assumptions change
allele and/or phenotype frequencies; examine how selection leads to evolutionary change; how secondary growth proceeds in woodyplants; and the linkage of predator-prey population cycles. Again, this was not
something I put in place overnight- my goal has been to develop/swap in 1-2
exercises per year. This gives me time to adequately develop the individual
exercises (and reduces my stress level). This involves talking to my
colleagues/grad students/undergrads about area where they see the greatest
difficulties, then brainstorming about what would help. I am also very
fortunate that Laurier has a great community of instructors and lots of
institutional support.
OK. This ended up being longer than I
expected, but I still think there is much more to discuss. Please feel free to
comment below, and I will do by best to answer any questions you may have! I
hope this has been of help!
TL
PS – If I can offer some other advice? Get
yourself a digital audio recorder and a small wired “lapel/lapellier
microphone, record all your lectures and put them immediately online your
course’s website. Not only does help with accessibility issues, but it helps
students out if they miss the occasional class, and/or if they can’t hear a key
moment because of a local noise in the classroom.
PPS- If you are a BI111 student who has
ended up here looking for more tips on how to do well in this class - remember to look at your learning objective
and (as always) read your syllabus!











{kind=link}